Sindromi poliendocrine

- Dettagli
- Categoria: Lezioni
- Pubblicato Sabato, 20 Luglio 2013 15:19
- Scritto da Paolo Purri
- Visite: 9177
 01. L'espressione clinica delle neoplasie multiple endocrine - sincrone o metacrone - si avvale delle conoscenze del sistema cosiddetto APUD, acronimo che raccoglie nelle iniziali le più comuni e caratteristiche proprietà citochimiche di una serie di cellule di origine neuroectodermica, capaci di una comune funzione : la sintesi e la secrezione di peptidi o amine ormonali. La definizione esplicita l'attività primaria di queste cellule, cioè il loro contenuto in amine derivanti dai processi di assunzione dei precursori di dopamina e 5-idrossitriptamina e successiva loro decarbossilazione per giungere al prodotto finale; caratteristica precipua di queste cellule è la metacromasia ed argirofilia, oltre all'alto contenuto di colinesterasi e di esterasi non-specifiche, nonchè gli elevati livelli di a-glicerofosfato-deidrogenasi mitocondriale. La microscopia elettronica, infine, rivela la presenza di granuli simili a quelli presenti nelle cellule endocrine. Cellule APUD del sistema neuroendocrino centrale si trovano nell'ipofisi, nella ghiandola pineale (epifisi) e nell'ipotalamo : * quelle ipofisarie secernono ACTH, somatotropina, prolattina e melanotropina; * quelle epifisarie secernono melatonina; * quelle ipotalamiche producono ossitocina, vasopressina, TRF (Thyrotropin-releasing factor), somatostatina (Somatotropin-release inhibiting factor), SRH (Somatotropin-releasing hormone), MRF (Melanotropin-releasing factor). Cellule APUD del sistema neuroendocrino periferico si trovano : * nel pancreas --> producono insulina, glucagone, somatostatina e polipeptide pancreatico (PP); * nello stomaco --> producono gastrina, glucagone e sostanza P; * nell'intestino --> producono motilina, enteroglucagone, secretina, somatostatina, sostanza P, CCK, GIP e VIP; * nella tiroide --> producono calcitonina; * nelle paratiroidi --> producono paratormone; * nel glomo carotideo (ormoni tiroidei e 5-idrossitriptamine), nella pelle (VIP), nel polmone (ADH = adiuretina) e nell'apparato uro-genitale. Le citate cellule APUD dimostrano una capacità multipotenziale nella produzione di peptidi ormonali che è alla base delle considerazioni che seguiranno nella trattazione delle patologie poliendocrine. |
 |
| 02. L'esordio delle sindromi poliendocrine come entità cliniche definite si verifica nel 1903 con il primo riscontro autoptico da parte di Erdheim della coesistenza di un adenoma eosinofilo ipofisario con una iperplasia paratiroidea; reperto confermato successivamente da Cushing e Davidoff nel 1927. Il crescente interesse scientifico porta nel 1939 alla prima ipotesi di una spiegazione genetica da parte di Rossier in seguito al riscontro di coesistenza di neoplasie delle paratiroidi e del pancreas insulare, unitamente a nefrolitiasi, in due sorelle di un nucleo familiare nel quale altri membri presentavano lesioni peptiche gastriche. Questo dato - confermato da Shelburn e Mc Laughlin nel 1945 e da Underdhal, Woolner e Black nel 1953 - spiana la strada al complesso capitolo delle sindromi poliendocrine, a partire da Wermer che nel 1954 ipotizza nel complesso di "adenomatosi delle ghiandole interne" riscontrato in più membri della stessa famiglia un meccanismo di trasmissione genetica con carattere autosomico dominante ad alta penetranza ed espressività variabile. Si tratta dunque di patologie neoplastiche che coinvolgono più ghiandole endocrine, sia in sindromi ben definite (diapositiva 6) che in complessi ad inquadramento non definito. Il comportamento biologico di queste neoplasie - spesso maligne - rende ragione della sostituzione del precedente acronimo MEA (multiple endocrine adenomatosis) con l'attuale MEN (multiple endocrine neoplasms). |
 03. |
 04. |
 |
| 05. |
 06. |
 |
| 07. |
 08. |
 |
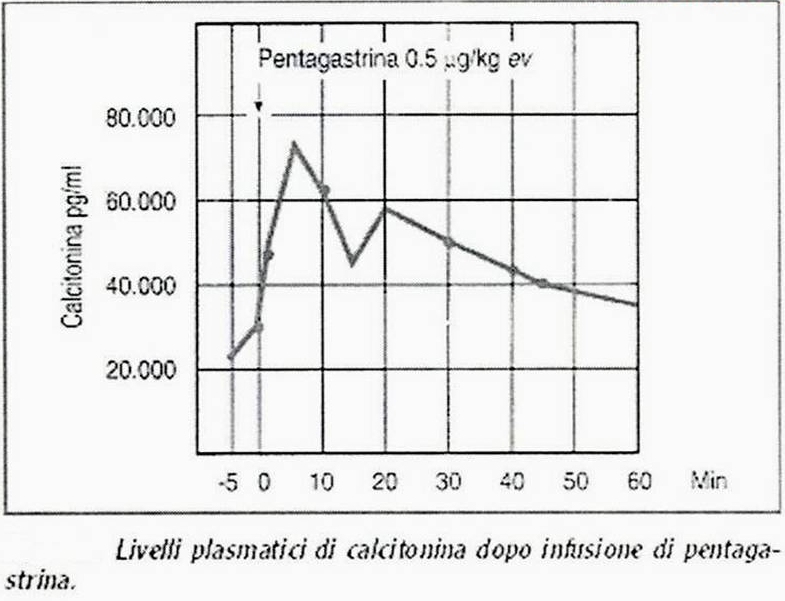
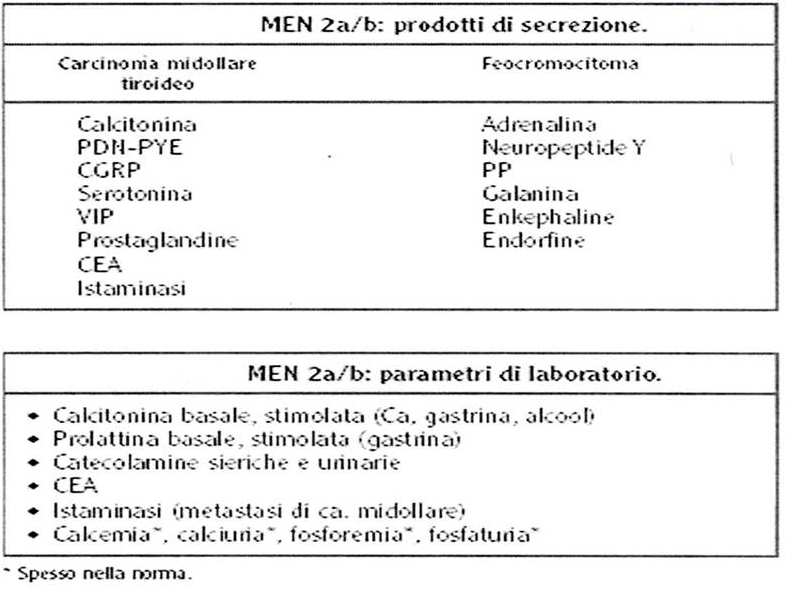
| 09. Una delle neoplasie ghiandolari più coinvolte nelle sindromi poliendocrine è certamente il carcinoma midollare della tiroide. A lungo considerato un carcinoma anaplastico, per la prima volta nel 1959 fu classificato da Hazard come una neoplasia con sue caratteristiche specifiche, sia oncogenetiche che biologiche. Il tumore origina, infatti, dalle cellule C della tiroide, disposte negli spazi interfollicolari ed immerse in uno stroma ad elevato contenuto in amiloide (elemento diagnostico, dal momento che è il solo tumore tiroideo a possederla); dette cellule sono produttrici di un ormone - la calcitonina, polipeptide composto da 32 aminoacidi - attivo sul metabolismo del calcio. Per questa loro posizione strutturale nella tiroide dette cellule sono definite parafollicolari o epifollicolari o cellule chiare; la loro pluripotenzialità funzionale è testimoniata dalla produzione - oltre alla calcitonina - di altre sostanze ad attività ormonale e non (diapositiva 9). Proprio per la sua origine da cellule indovate negli spazi interfollicolari non possiede capacità di captare lo iodio radiattivo, mentre mostra una spiccata tendenza a concentrare un tracciante (MIBG) che si fissa elettivamente al tessuto cromaffine del feocromocitoma e di altre neoplasie di origine neoroectodermica; ed è questa caratteristica che ha consentito di inquadrare il tumore nei casi di familiarità con ereditarietà trasmessa con carattere autosomico dominante (sindrome di Sipple). La neoplasia isolata non mostra predilezione riguardo al sesso, così come tutte le età possono essere colpite; la forma familiare, invece, predilige il sesso femminile ed è più frequente nella 2^-3^ decade di vita. L'aspetto macroscopico nelle forme isolate è quello di un nodulo unico, di dimensioni assai variabili (da pochi millimetri fino a 10 centimetri); la multicentricità e la bilateralità sono invece tipiche delle forme familiari; caratteristica comune è l'aspetto microscopico nel quale spicca la presenza di amiloide, strettamente diagnostica di questa forma neoplastica tiroidea. Pur con una crescita lenta, il tumore diffonde relativamente precocemente ai linfonodi del collo e del mediastino superiore, più tardivamente al polmone, al fegato ed alle ossa (lesioni frequentemente osteoblastiche); caratteristica delle metastasi è la conservazione dell'attività secretoria della lesioni di partenza (cosa che viene sfruttata nel follow-up dei pazienti tiroidectomizzati). La clinica evidenzia la pluripotenzialità secretoria delle cellule neoplastiche: infatti è presente diarrea (in almeno il 30% dei casi) ed ipermotilità intestinale quali espressioni della produzione di serotonina, calcitonina, prostaglandine E2 ed F2a; alla produzione di ACTH è correlata la sindrome di Cushing presente in almeno 5% dei carcinomi midollari tiroidei (gli unici tra tutte le neoplasie tiroidee a mostrare questa associazione clinica). Spesso si accompagnano disfagia ed alterazione del timbro e del tono della voce (per invasione neoplastica dell'esofago e della trachea); già all'atto della diagnosi si dimostra l'impegno delle stazioni linfonodali regionali (circa 25% dei casi), con reperto ancor più elevato (fino a 75% dei casi) all'atto operatorio. La diagnosi è agevolata dal riscontro di elevati tassi circolanti di calcitonina, incrementati dall'infusione di calcio o dall'assunzione di alcolici. Per le caratteristiche stesse del tumore, che ne limitano le opportunità terapeutiche alternative (diapositiva 15), il trattamento non può essere che chirurgico: la tiroidectomia totale con linfoadenectomia latero-cervicale è il procedimento di elezione, in considerazione delle caratteristiche anatomiche (multifocalità) e biologiche (pluripotenzialità cellulare); tuttavia per lesioni plurifocali (inquadrate in una sindrome poliendocrina) la frequente disposizione polare superiore delle cellule C nell'ambito tiroideo consente una procedura alternativa di tiroidectomia subtotale con risparmio di una modesta area tissutale della base del lobo controlaterale a quello interamente asportato. A questa scelta chirurgica - decisamente meno aggressiva dell'exeresi radicale - contribuisce la conoscenza di una minore aggressività biologica dei carcinomi midollari plurifocali, con una prognosi decisamente migliore rispetto alle forme uniche (fatta eccezione per quelli inquadrati in una MEN 2b). |
 |
| 10. |
 |
| 11. |
 12. |
 |
| 13. |
 14. |
 |
| 15. |
 |
| 16. Altro comune componente delle sindromi poliendocrine è il feocromocitoma, neoplasia che origina dalle cellule cromaffini della midollare del surrene. Il tumore secerne catecolamine ad azione ipertensivante, sia di tipo accessionale (episodico) che di tipo intermittente o stabile; spesso una crisi ipertensiva parossistica può determinare la morte in soggetti affetti dalla neoplasia. L'identificazione precoce del tumore e la rapida messa in opera di tutti i presidi atti a contenere gli effetti dell'attività secretoria delle cellule neoplastiche consentono di limitare sensibilmente la mortalità ed incrementare in modo significativo gli indici prognostici. Alla caratteristica produzione di catecolamine il tumore associa la secrezione di ACTH (sindrome di Cushing concomitante), oltre a somatostatina, ossitocina, vasopressina e calcitonina (tutti, però, in quantità insufficiente a determinare una sindrome clinica dipendente). La bilateralità della neoplasia è caratteristica della sindrome di Sipple (MEN 2a) e di Gorlin-Schimke (MEN 2b), mentre in forma isolata il tumore è monolaterale (associato ad iperplasia controlaterale). Le lesioni extra-surrenaliche coinvolgono il collo, il torace e l'organo di Zuckerkandl. L'incidenza di malignità della neoplasia è discretamente bassa (inferiore al 10% nelle casistiche più accreditate); spesso si riconoscono segni istologici di malignità, incluso l'infiltrazione venosa nell'area neoplastica, senza tuttavia dimostrazione di un vero potenziale maligno conseguente; in certi casi si riconosce una degenerazione maligna di alcuni focolai multicentrici benigni. La variabilità di condizioni descritte induce alla attribuzione di unico criterio di malignità alla chiara evidenza di metastasi in sedi nelle quali è inusuale la presenza di cellule cromaffini; sulla base di questo criterio l'indice di malignità risulta compreso tra 2,5% e 15%. Le metastasi sono frequentemente funzionanti, determinando una ricomparsa della sintomatologia tipica parallelamente alla diffusione delle lesioni primitive; spesso, anzi, esse precedono l'evidenza delle lesioni primarie in virtù della loro capacità di produzione dei derivati metabolici della dopamina, dell'epinefrina e della nor-epinefrina. Nella forma isolata il tumore è più frequente a carico del surrene destro; le dimensioni macroscopiche oscillano tra 3 e 5 cm. Le lesioni di dimensioni maggiori presentano aree cistiche intercalate a zone necrotiche nel loro contesto, con frequenti calcificazioni. Una sottile banda di tessuto fibroso circoscrive il tessuto neoplastico, isolandolo dal corticosurrene, che tuttavia può a volte risultare invaso per pregnazione vascolare delle componenti benigne associate. Il quadro clinico (diapositiva 21) è dominato da sintomi prodromici aspecifici - quali cefalea, nausea, vomito, dolori addominali, sudarozione e palpitazioni - cui segue la manifestazione patognomonica della patologia : l'ipertensione, sia di tipo stabile e continuo che di tipo intermittente e parossistico, spesso scatenata da stress o da procedure diagnostiche (radiologiche contrastografiche) o da assunzioni di farmaci (chemioterapici ed antidepressivi). Il sospetto diagnostico trova conferme nelle indagini di laboratorio e nei tests di stimolazione (istamina, tiramina, glucagone) e soppressione (fentolamina, clonidina), con risultati fortemente indicativi della presenza di una neoplasia piuttosto che di una iperplasia medullo-surrenalica. Al riconoscimento dell'esistenza della lesione segue la necessaria sua localizzazione: al raggiungimento di questo obiettivo concorrono debitamente sia TAC, RNM e scintigrafia con MIBG che l'arteriografia selettiva e - più ancora - la flebografia retrograda surrenalica (con raccolta di campioni ematici per la determinazione delle catecolamine specifiche). La strategia terapeutica - essenzialmente chirurgica - è articolata e complessa per la delicatezza delle condizioni cliniche del paziente, spesso ai limiti del rischio. La necessità di contenere i valori pressori entro limiti accettabili e di dominare le frequenti aritmie associate impongono un trattamento medico pre-operatorio ed intra-operatorio mediante somministrazione di a- e ß-bloccanti recettoriali adrenergici; detto trattamento risulta utile anche in presenza di metastasi funzionanti o per pazienti che rifiutino un trattamento chirurgico. Le procedure operatorie da porre in essere sono riferite alla presenza di una lesione isolata monolaterale o coinvolta in una sindrome MEN (e pertanto spesso bilaterale); in quest'ultimo caso - in cui la lesione neoplastica in un surrene si accompagna ad iperplasia medullosurrenalica controlaterale - una surrenalectomia monolaterale associata ad una resezione subtotale controlaterale può preservare la funzionalità ghiandolare residua ed evitare una terapia sostitutiva permanente. In ogni caso la via di accesso preferibile è quella anteriore addominale per una accurata esplorazione alla ricerca di eventuali tumori ectopici; il controllo pressorio intraoperatorio è imperativo al fine di valutare il successo terapeutico della procedura attuata, in quanto l'eventuale persistenza di livelli pressori elevati dopo la legatura della vena surrenalica è indicativa della sussistenza di una ulteriore localizzazione neoplastica che deve necessariamente essere ricercata. Ai soli casi di inoperabilità sono riservate procedure di embolizzazione o trattamenti radianti. La netta differenza di sopravvivenza a 5 anni tra forme benigne e maligne (90% contro 40% rispettivamente), unitamente alle oggettive difficoltà di definizione in fase diagnostica della natura benigna o maligna della lesione, rende ragione dell'opportunità di un prolungato follow-up di questi pazienti, un 10% dei quali presenta recidiva anche a dispetto di trattamenti decisamente corretti. |
 |
| 17. |
 |
| 18. |
 |
| 19. |
 20. |
 21. |
 22. |
 23. |
 24. |
 25. |
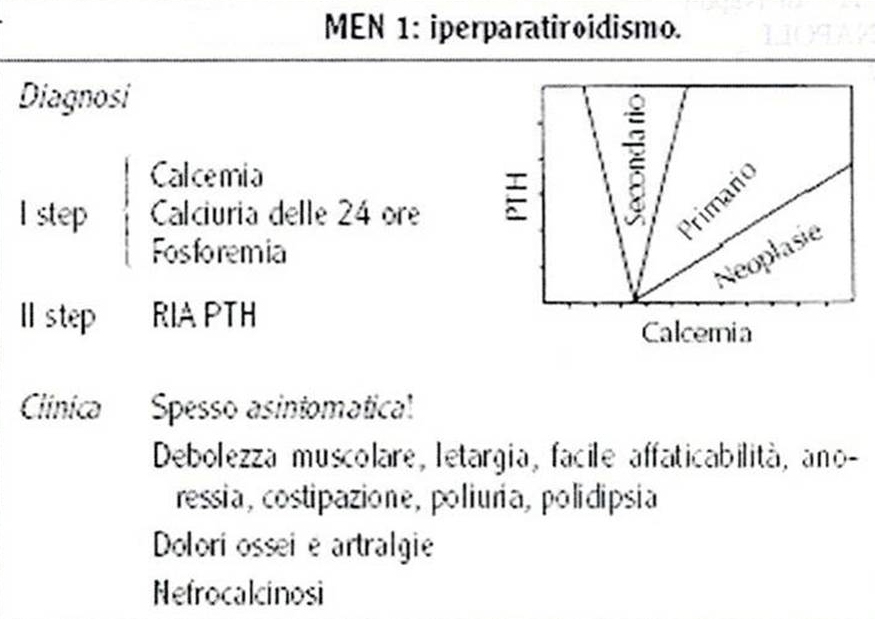
 26. Una delle componenti della sindrome di Wermer (MEN 1) è l'adenoma ipofosario, descritto per la prima volta da Cushing e distinto in cromofobo (50% circa), eosinofilo e basofilo in base alla componente cellulare che lo contraddistingue ed alle rispettive capacità secretorie di sostanze ormonali. Tale distinzione grossolana nei tre tipi cellulari citati è stata superata da una più recente e dettagliata in 7 sottotipi cellulari (a, ß, ß1, ß2, d1, d2, y) mantenendo la qualifica di cellula cromofoba alla sola cellula piccola o staminale o in fase dì riposo (la cui quantità risulta pertanto inferiore al 50% indicato in precedenza). Caratteristicamente i tumori non secernenti adenoipofisari (adenomi cromofobi) si segnalano per l'espansione nell'ambito della sella turcica e compressione delle unità cellulari secernenti confinanti, provocandone una riduzione della funzione; in ordine decrescente ne sono coinvolti l'ormone della crescita, le gonodotropine, l'ormone tireotropo e da ultimo l'ACTH. Queste funzioni sono generalmente soppresse in sequenza, così che una deficienza surrenalica è preceduta da una ipofunzione tiroidea e gonadica; mentre un quadro di diabete insipido è di raro riscontro. L'espansione verso l'alto, oltre i confini della sella turcica, comporta fenomeni compressivi sul chiasma ottico e la tipica emianopsia bitemporale; mentre l'estensione verso il terzo ventricolo determina idrocefalo e deficit funzionali ipotalamici. Molto più rara l'espansione laterale, con compressione diretta sui nervi cranici entro il seno cavernoso (oculomotore, trocleare, abducente e primo ramo di divisione del trigemino); più spesso la compromissione riguarda il loro supporto vascolare ("apoplessia pituiria") con conseguente deficit funzionale. Un quadro clinico direttamente dipendente dall'attività funzionale della neoplasia si registra per adenomi che originano dalle componenti cellulari cromofile dell'adenoipofisi: adenomi eosinofili (secernenti prolattina o GH) e basofili (secernenti ACTH). Il più frequente nella MEN 1 (70% di incidenza) è certamente il prolattinoma, tumore con capacità di accrescimento fino a dimensioni cospicue e slargamento della sella turcica; non mancano, tuttavia, forme di dimensioni assai contenute (microadenomi), senza modifiche volumetriche della sella e pertanto di difficile identificazione radiologica pre-operatoria. Aldilà delle manifestazioni da compressione, tipiche di tutti gli adenomi ipofisari, la sintomatologia specifica connessa con l'iperincrezione di prolattina contempla amenorrea, galattorrea, infertilità nei soggetti di sesso femminile ed impotenza nei maschi. Dato significativo è che l'eccesso di liberazione di prolattina non risulta potenzialmente letale come quello di somatotropina o di ACTH. Secondo in ordine di frequenza nella MEN 1 è l'adenoma produttore dell'ormone della crescita, responsabile di acromegalia se insorge in età adulta e di gigantismo se si manifesta in età adolescenziale. L'effetto dell'iperproduzione ormonale si esplica sulle ossa (con accrescimento eccessivo), sulle masse muscolari (con incremento volumetrico e ponderale), sul metabolismo glicidico (con aumento dei livelli glicemici ematici); caratteristico e diagnostico è l'aumento di volume delle estremità (mani e piedi) unitamente alle ossa del massiccio facciale (facies acromegalica). L'incremento volumetrico coinvolge anche gli organi, in particolare il cuore con il risultato di compromettere la funzione valvolare e del miocardio in toto, con alto rischio di mortalità. La crescita intrasellare del tumore - di per sè già causa di ipofunzione delle altre cellule secernenti dell'adenoipofisi - spesso sconfina oltre i limiti della struttura ossea, comportando la comparsa di turbe visive ed idrocefalo. Una particolare attenzione deve essere rivolta al fatto che a fronte di microadenomi eosinofili GH-secretori la maggior parte dei tumori responsabili di acromegalia sono adenomi cromofobi con granuli di ormone nel citoplasma cellulare (cellule potenzialmente funzionanti). Per il citato rischio di mortalità connesso con gli esiti miocardiopatici enunciati è chiaro che l'obiettivo del trattamento deve essere rivolto all'abbassamento del livello dell'ormone circolante: allo scopo la strategia terapeutica si avvale della semplice asportazione dell'area neoplastica per i microadenomi e della ipofisectomia per lesioni maggiori (con effetti sulle altre componenti adenoipofisarie). La radioterapia mirata risulta efficace per il contenimento dell'espansione neoplastica rendendo più agevole l'approccio chirurgico. Terzo in ordine di frequenza - e peraltro abbastanza raro componente di una MEN 1 - è l'adenoma ipofisario produttore di ACTH. Si tratta di un adenoma basofilo, la cui ipersecrezione ormonale si riflette nell'ipersecrezione di steroidi ormonali da parte del cortico-surrene. E' bene tenere presente che non tutti i pazienti con elevati livelli plasmatici di corticosteroidi devono questa condizione ad un tumore ipofisario produttore di ACTH; l'accertamento della dipendenza dell'asse ipofisi-surrene deve essere demandato ai risultati dei tests di stimolazione/inibizione. Generalmente il tumore ipofisario è piccolo e non modifica la struttura della sella turcica; per questo motivo un aspetto normale della sella non esclude la presenza di una neoplasia funzionante ipofisaria per la cui conferma è imperativa una indagine ormonale a largo spettro. La conoscenza del ruolo dell'asse ipotalamo-ipofisario a proposito dell'interrelazione tra CRF (fattore di liberazione della corticotropina, denominato anche corticoliberina) ed ACTH fa comprendere il complesso meccanismo di azione dell'adrenocorticotropina sulla corticale del surrene per la produzione del cortisolo in condizioni normali; l'alterazione del meccanismo feed-back cortisolo-ACTH - con elevata produzione di CRF ed ACTH a dispetto di elevati tassi di cortisolo circolante - è alla base della patologia caratterizzata dal complesso sintomatico illustrato nelle diapositive 36 e 37. La complessità del quadro anatomo-clinico obbliga ad una particolare accuratezza diagnostica, specie in ragione delle molteplici sedi di adenomi funzionanti ectopici (diapositiva 35); allo scopo, oltre all'ausilio fornito dallo studio radiografico della sella turcica (x-grafia tradizionale del cranio, TAC e RNM), contribuiscono la determinazione della curva giornaliera del cortisolo ematico ed urinario ed i tests di stimolazione/soppressione specifici per una diagnostica differenziale di lesioni a carico del corticosurrene (diapositiva 38), capaci di indirizzare verso una patologia surrenalica primitiva o mediata. Alla luce delle informazioni acquisite il trattamento terapeutico è necessariamente modulato in ragione del meccanismo patogenetico della sindrome dipendente: così dinanzi ad un Cushing dipendente da adenoma ipofisario la priorità di intervento sulla neoplasia piuttosto che sull'iperplasia surrenalica risulta imperativa. |
 27. |
 28. |
 29. |
 30. |
 31. |
 32. |
 33. |
 34. |
 35. |
 36. |
 37. |
 38. |
 39. |
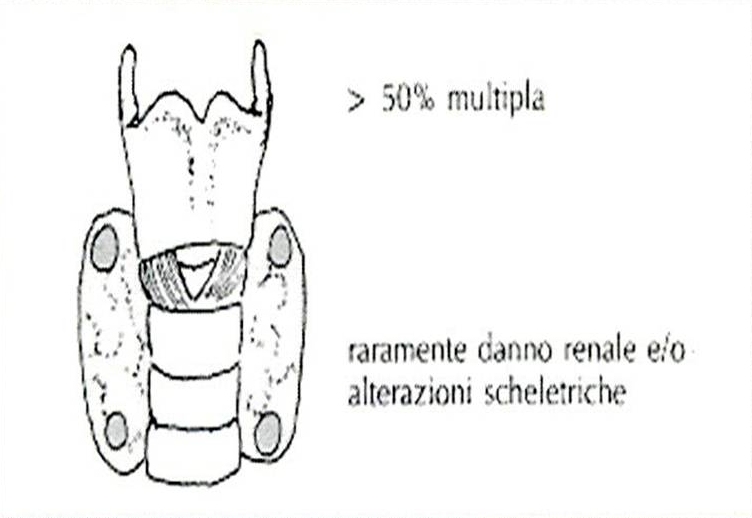
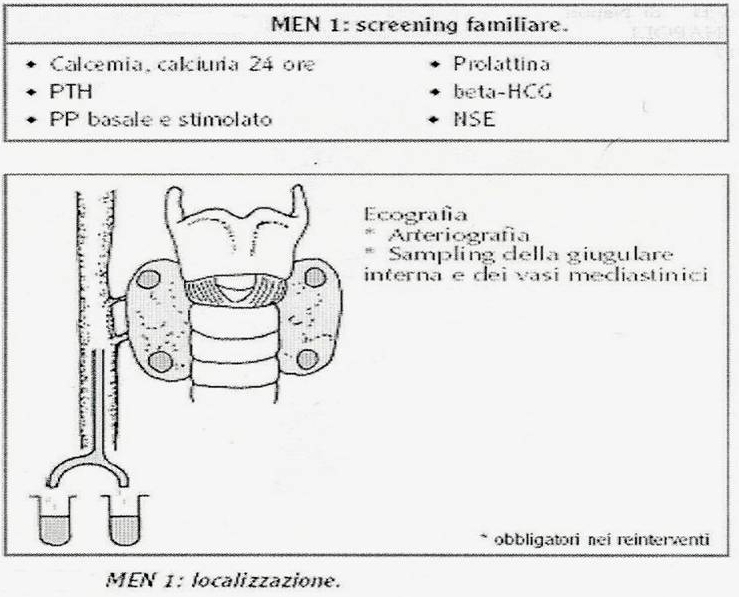
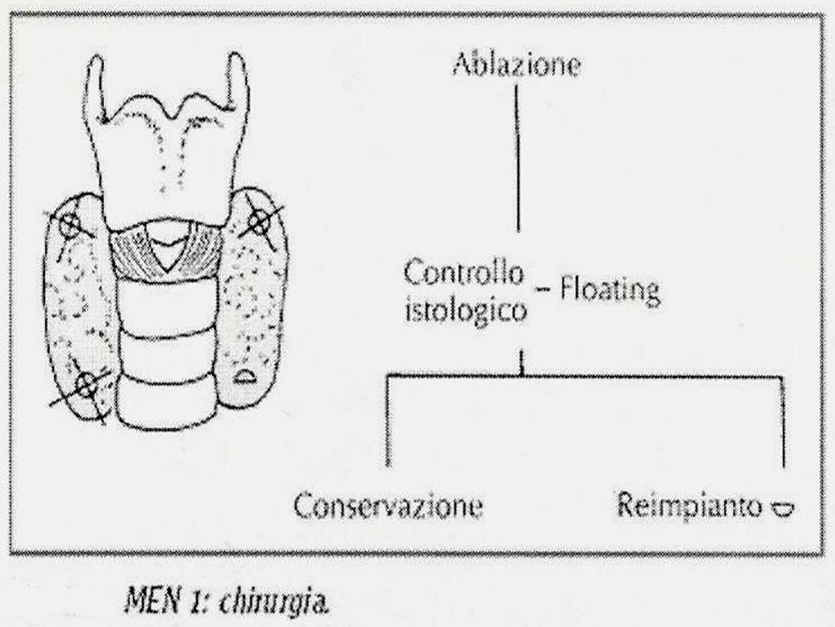
 40. Quanto premesso rende più agevole la comprensione dei due complessi clinici principali nell'ambito delle sindromi poliendocrine: le sindrome di Wermer e la sindrome di Sipple. La sindrome di Wermer consiste nell'associazione di adenomi ipofisario, paratiroideo ed insulo-pancreatico; frequentemente si associano adenomi del surrene e della tiroide, oltre a carcinoidi (intestinali e bronchiali). In una larga percentuale di casi (oltre 85%) si riscontrano lipomi multipli e fibromi o angiofibromi facciali, con una patogenesi assimilabile a quella delle componenti endocrine della sindrome e riferibile ad una mutazione allelica del cromosoma 11q13. Le paratiroidi sono più frequentemente coinvolte nella sindrome e l'iperparatiroidismo è evidente mediamente in 90% dei casi; sebbene calcemia, fosforemia e paratormone risultino costantemente elevati, la sintomatologia manifesta può essere più facilmente riferita a neoplasie dell'ipofisi o del pancreas dal momento che danni scheletrici o renali sono piuttosto rari (ancorchè gravi). Un adenoma paratiroideo si riscontra in circa il 40% dei casi di MEN 1, mentre per i restanti domina il quadro dell'iperplasia ghiandolare; comunque il coinvolgimento è più frequentemente multiplo (oltre il 50% dei casi) e richiama l'attenzione sulla necessità di valutazione anche di altre ghiandole endocrine nei casi di familiarità della manifestazione clinica. Il coinvolgimento insulare pancreatico si riscontra in una media del 75% dei casi (con picchi compresi tra 65 e 80%), con una larga percentuale (almeno 65%) rappresentata da adenomi del tipo non-ß-cellulare, con prevalente secrezione di gastrina (50% dei pazienti con MEN 1 presentano una storia di ulcera peptica) o di insulina (15%); più rari gli adenomi secernenti glucagone, ACTH, VIP, ADH, prostaglandine, somatostatina e serotonina. Gli insulinomi non-funzionanti risultano maligni in 1/3 dei casi. Una nota di particolare interesse riguarda proprio la frequenza di lesioni ulcerative peptiche nell'ambito della MEN 1: infatti, anche se in almeno 2/3 dei casi si tratta di ulcere complicate - come quelle riscontrate nella Zollinger-Ellison dipendente da un tumore non-familiare pancreatico produttore di gastrina - solo il 3% dei casi sporadici di Z-E presentano una positiva storia familiare con coinvolgimento di altre strutture endocrine, mentre un 20% di quelli senza familiarità accertata mostra un coinvolgimento di altre ghiandole endocrine (paratiroidi 65% e adenoipofisi 35%). Secondo in ordine di frequenza tra i tumori insulari pancreatici coinvolti in una MEN 1, l'insulinoma pancreatico è nel 35% dei casi un adenoma ß-cellulare e solo nel 3% un adenoma misto (di tipo ß- e non-ß-cellulare), con il primo del tutto simile agli insulinomi sporadici anche nel corredo sintomatologico ben noto (diapositiva 47). Circa il 50% dei pazienti con una sindrome di Wermer presentano un adenoma adenoipofisario, in prevalenza di tipo cromofobo con chiari segni di ipopituitarismo ed in misura minore di tipo eosinofilo o misto (eosinofilo, basofilo e cromofobo); il quadro clinico è in relazione con l'ormone secreto (come riportato in diapositiva 51) unitamente alle manifestazioni proprie della compressione dovuta all'espansione neoplastica (diplopia ed emianopsie). Un significato particolare assumono i casi occasionali di associazione di tumori tiroidei con adenomi corticosurrenali; infatti in assenza di una lesione neoplastica ipofisaria le manifestazioni cliniche di una sindrome di Cushing sono del tutto assenti, mentre la tiroide è rappresentata con adenomi isolati e molto meno frequentemente con carcinomi o espressioni di tireotossicosi. La variabilità di ulteriori componenti patologiche è sinteticamente rappresentata nella diapositiva 54, a conferma di una sindrome con diverse espressioni cliniche in riferimento agli effetti funzionali delle sostanze ormonali messe in circolo. Le procedure diagnostiche e le strategie di trattamento per i singoli componenti la sindrome non si discostano da quelle messe in atto per le neoplasie sporadiche o isolate; è essenziale, tuttavia - in considerazione della dipendenza genetica dell'associazione - lo screening dettagliato nei casi di familiarità, da ripetersi con regolarità (almeno una volta l'anno). Infatti è diffusamente riconosciuta l'esistenza di un difetto genetico all'origine del coinvolgimento ipofisario, pancreatico e paratiroideo nei casi di familiarità: una trasmissione autosomica dominante con alta penetranza, ma ovviamente non uniforme dal momento che tutte e tre le ghiandole non sono costantemente coinvolte. Il supporto radiologico per immagini e lo studio angiografico e scintigrafico (diapositiva 56) sono di grande utilità per l'identificazione e localizzazione delle lesioni, nonchè per la programmazione terapeutica: la plurifocalità delle lesioni a carico delle paratiroidi suggerisce l'ablazione delle quattro ghiandole accessibili con reimpianto parziale di una di esse in una area della loggia muscolare dell'avambraccio (per limitare una sindrome da ipoparatiroidismo); la frequente malignità delle lesioni pancreatiche, spesso causa di morte, indica l'opportunità di una pancreasectomia totale (o eventualmente sub-totale, con radicalizzazione mediante a-interferone e octreotide); ai tumori ipofisari si riserva una ablazione modulata (totale o parziale, secondo necessità) condotta con accesso frontale o per via trans-sfenoidale. La terapia medica (diapositiva 60) si riserva ai prolattinomi, agli insulomi non-ß-cellulari ed agli insulinomi benigni, mentre gli inibitori di pompa sono opportuni nei casi di eccessiva produzione acida gastrica da iperproduzione gastrinica (ovviamente dopo trattamento chirurgico delle eventuali perforazioni). La presenza eventuale di focolai secondari epatici si giova di procedure di chemioembolizzazione, alcolizzazione, crioablazione o necrotizzazione a radiofrequenza. |
 41. |
 42. |
 43. |
 44. |
 45. |
 46. |
 47. |
 48. |
 49. |
 50. |
 51. |
 52. |
 53. |
 54. |
 55. |
 56. |
 57. |
 58. |
 59. |
 60. |
 61. |
 62. |
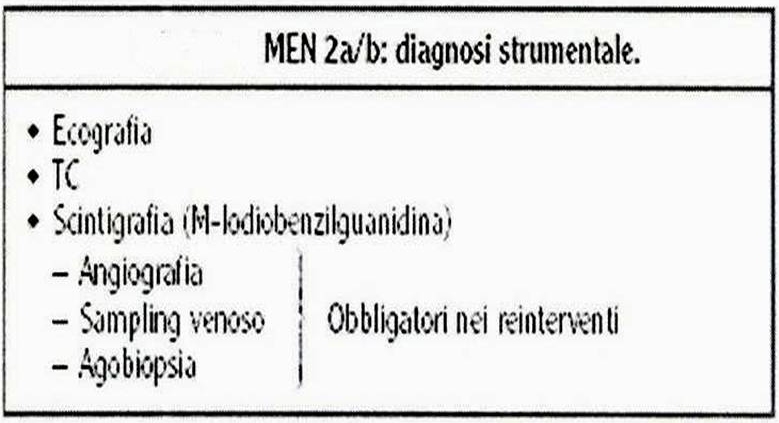
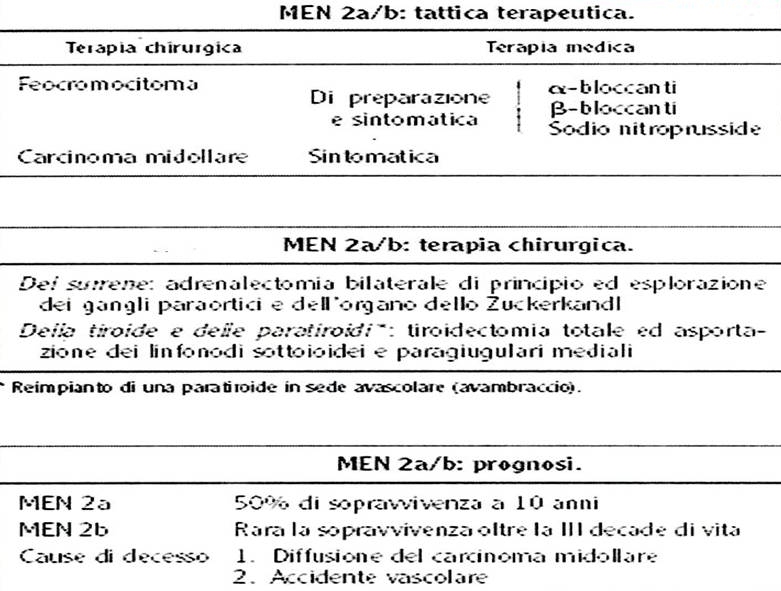
 63. La sindrome di Sipple (MEN 2) è caratterizzata dall'associazione di carcinoma midollare della tiroide (componente costante), feocromocitoma (40% dei casi e spesso bilaterale) e iperplasia o adenoma paratiroideo. E' una sindrome familiare con una alta variabilità di penetranza per le diverse componenti ghiandolari; l'epoca di insorgenza delle singole lesioni non è simultanea, così come l'interessamento ghiandolare nei casi di bilateralità. Il carcinoma midollare tiroideo è bilaterale nel 70-80% di casi di sindrome familiare e solo nel 30% delle forme sporadiche; il feocromocitoma è bilaterale nel 50-80% dei casi di sindrome familiare e solo nel 5% delle forme sporadiche. Nella variante 2a la "notalgia parestesica" (diapositiva 66) rappresenta un indice diagnostico di rilievo per la sua specificità, unitamente all'eventuale presenza di ipertrofia dei nervi corneali; nella variante MEN 2b spicca il caratteristico "habitus marfanoide" - con multipli neuromi mucosi, labbra rigonfie, miotonia ed anomalie scheletriche; non sono presenti l'aneurisma dissecante aortico e le disfunzioni valvolari cardiache, tipici riscontri della sindrome di Marfan. Tale quadro può manifestarsi già alla nascita, ma non vi è assoluta la concomitanza del carcinoma tiroideo in questa fase della vita. Le componenti ghiandolari sono le stesse per entrambe le varianti, anche se l'interessamento paratiroideo è molto più sporadico (0,5%) nella MEN 2b; la neoplasia tiroidea è più precoce, più aggressiva e spesso multifocale nella MEN 2b, mentre le lesioni surrenaliche sono raramente maligne in entrambe le forme familiari. Una terza variante della MEN 2 è la FMTC (carcinoma midollare familiare tiroideo) nella quale la neoplasia riconosce una insorgenza su base ereditaria del tipo autosomico dominante e non è associata ad altri tumori ghiandolari; tipicamente la lesione mostra una aggressività inferiore a quella riscontrata nelle forme sporadiche o inquadrate in una MEN 2. Fattore comune alla tre varianti descritte è una mutazione germinale a livello del protogene RET, prevalentemente nella porzione extracellulare per i casi di FMTC e nelle sequenze intracellulari nei casi di MEN 2a e 2b; differenti tipi di mutazione caratterizzano una diversa penetranza della sindrome. Per una migliore comprensione è utile ricordare che il gene RET (rearranged during transfection) è un proto-oncogene manifesto esclusivamente nelle cellule di derivazione neuroectodermica (come le cellule C della tiroide); localizzato sul cromosoma 10 (10q11.2), codifica per una proteina chinasi di superficie e funziona da recettore per una serie di proteine della famiglia del GDNF (fattore di crescita delle cellule gliali). L'attivazione del recettore si realizza mediante un processo di dimerizzazione dopo il legame con un corecettore del tipo GFRa. Il recettore Ret ha un dominio CAL (caderine-like) e un dominio ricco in cisteine nella porzione extracellulare, e due domini TK (tirosinchinasi) nel versante citoplasmatico. Nella forma mutata (Ret/ptc) il gene, a trasmissione autosomica dominante, è correlato all'insorgenza di alcune neoplasie, tra cui le MEN e il carcinoma midollare familiare della tiroide (FMTC). Le mutazioni più comuni della proteina RET correlate a queste manifestazioni tumorali sono: • MEN 2A: sostituzione di una cisteina in posizione 634 • FMTC: sostituzioni di cisteine in altre posizioni e sostituzioni ai domini TK • MEN 2B: sostituzione in posizione 918 di una metionina con una treonina. L'importanza di queste conoscenze spiega perchè tutti i soggetti appartenenti a famiglie con casi di MEN 2 devono essere sottoposti a screening genetico alla nascita: l'eventuale riscontro di una mutazione a rischio suggerisce l'opportunità di procedere ad una tiroidectomia e paritiroidectomia totale entro il primo anno di vita nei casi di MEN 2b ed entro i primi 5 aa. nei casi di MEN 2a e di FMTC; lo screening è consigliabile anche nei casi di carcinoma midollare solitario. L'accertamento diagnostico, mediante metodiche di screening sia di laboratorio che radiologico (diapositive 71, 73, 76, 77), fornisce informazioni dettagliate sulle diverse componenti ghiandolari delle varianti descritte e sulla localizzazione delle neoplasie nell'ambito delle stesse; ciò risulta di grande efficacia per una corretta programmazione terapeutica ed un elevato risultato prognostico. I capisaldi del progetto terapeutico sono così sintetizzati: a) timing prioritario all'asportazione del feocromocitoma nell'associazione di questo con un carcinoma tiroideo; b) atteggiamento esplorativo debitamente aggressivo avverso il feocromocitoma per la sua frequente multifocalità (ingiustificata una surrenalectomia bilaterale di principio); c) tiroidea totale, unitamente alle stazioni linfonodali satelliti di primo e secondo livello; d) ablazione sub-totale delle paratiroidi con recupero di parte di una di esse per l'impianto in una loggia muscolare dell'avambraccio. Nonostante la grande somiglianza di espressione clinica della MEN 2a e 2b, il discorso prognostico si discosta notevolmente e la sopravvivenza mostra indici a distanza assai diversi: la particolare aggressività delle neoplasie componenti la sindrome MEN 2b, unitamente alla precocità di insorgenza, fanno sì che a fronte di una sopravvivenza a 10 anni intorno al 50% per soggetti affetti da MEN 2a ben pochi tra quelli affetti da MEN 2b superano la 3^ decade di vita. |
 64. |
 65. |
 66. |
 67. |
 68. |
 69. |
 70. |
 71. |
 72. |
 73. |
 74. |
 75. |
 76. |
 77. |
 78. |
 79. |
 80. |
 81. |
 82. Una analisi della letteratura sull'argomento fa comprendere quanto la grande variabilità di associazioni tra neoplasie o quadri di iperplasie ghiandolare renda difficile una classificazione unitaria delle sindromi polendocrine; tanto che più correttamente si potrebbe definire il complesso delle MEN una costellazione di "sindromi cliniche endocrine" variamente concomitanti o associate tra loro. Aldilà delle forme di maggior frequenza fin qui riportate esistono, tuttavia, alcune altre meno rappresentate, come l'associazione <<carcinoide duodenale -feocromocitoma - neurofibromatosi>> e quella <<tumore insulare pancreatico o feocrocitoma ereditario - sindrome Von Hippel-Lindau>>. Nella prima, generalmente scoperta accidentalmente, il carcinoide mostra spesso positività alla somatostatina; pertanto le manifestazioni cliniche rispecchiano gli effetti locali e sistemici della secrezione ormonale: costipazione, malassorbimento, diarrea e soprattutto ittero (per la spiccata tendenza litogenica biliare con possibili ostruzioni duttali). La neurofibromatosi tipo 1 di Von Recklinghausen associata denuncia una dipendenza genetica della patologia con trasmissione autosomica dominante, nella quale è riconosciuta l'azione di un gene onco-soppressore (NF1 localizzato sul cromosoma 17) che codifica una proteina (neurofibrina) coinvolta nel controllo negativo della proliferazione cellulare. Nella seconda è dominante l'espressione di una sindrome di Von Hippel-Lindau, malattia a trasmissione autosomica dominante associata a varie neoplasie. La causa di questa malattia è una mutazione di un gene localizzato sul braccio corto del terzo cromosoma, mappato in 3p25-26, il quale codifica per la proteina VHL. A seconda del genotipo del soggetto si riscontrano quadri clinici differenti. La malattia è suddivisibile in 2 tipi: il tipo 1 senza associazione con feocromocitoma e il tipo 2 che invece la manifesta. Nel primo caso la proteina spesso è mutata, ma presente; nel secondo caso invece è assente. Inoltre il tipo 2 è diviso ulteriormente in 3 sottotipi: A, con tumore a cellule renali; B, con associato emangioblastoma; C, con il solo feocromocitoma. Sono anche riscontrati tumori endocrini (specie pancreatici), cisti renali (dovute all'edema da neoangiogenesi) ed altre manifestazioni neoplastiche. La diagnosi è indirizzata alla ricerca delle alterazioni del gene VHL e deve essere guidata dalla clinica con la quale la patologia si manifesta; la terapia è necessariamente chirurgica, con procedure specifiche per le diverse associazioni neoplastiche presenti. Un'ultima sindrome poliendocrina, anch'essa probabilmente trasmessa con tratto autosomico dominante, è quella che vede il concorso di carcinoma midollale multicentrico tiroideo, iperplasia paratiroidea e adenoma ipofisario con paragangliomi carotidei bilaterali, leiomiomi gastrici ed amiloidosi sistemica. |
 83. |
 84. |
 85. |
BIBLIOGRAFIA
Beckers A., Aaltonen L.A., Daly A..F, Karhu A. : Familial isolated pituitary adenomas (FIPA) and the pituitary adenoma predisposition due to mutations in the aryl hydrocarbon receptor interacting protein (AIP) gene. Endocr. Rev. 2013 : Apr. 34(2), 239-77.
Brandi M.L., Gagel R.F., Angeli A., et al. : Guidelines for diagnosis and therapy of MEN type 1 and type 2.J. Clin. Endocrinol. Metab. 2001: 86 (12), 5658-71.
Chu X., Gao X., Jansson L., Quach M., Skogseid B., Barbu A. : Multiple Microvascular Alterations in Pancreatic Islets and Neuroendocrine Tumors of a Men1 Mouse Model. Am. J. Pathol. 2013: Apr. 12.
Cuenca-Cuenca J.I., Marín-Oyaga V.A., Borrego-Dorado I., Navarro-González E., Martos-Martínez J.M., Vázquez-Albertino R. : (123)I-MIBG, (18)F-DOPA and (18)F-FDG in a patient with MEN2 syndrome and recurrent pheochromocytoma. Rev. Esp. Med. Nucl. Imagen Mol. 2013: Feb 20.
Cui Q., Wang W., Fu Z., Shao X., Zhang Z., Zhang M., Ju X., Wang K., Chen J., Zhou H. : Integrated DNA-based/biochemical screening for early diagnosis of multiple endocrine neoplasia type 2A (MEN2A). J. Biomed. Res. 2013: Mar. 27(2), 145-50.
Colao A., Ferone D., Marzullo P., Lombardi, G. : Systemic complications of acromegaly: epidemiology, pathogenesis and management.Endocrine Review 2004: 25,102-152.
Deng W., Lan L., Zuo Q.Y., Wang H.D., Bai N., Ding Y.:Genetic screening in families with multipleendocrine neoplasia type 2A for possible medullary thyroid cancer. Chin. Med. J. 2013: Apr.126(8),1599-600.
Gadelha M.R., Une K.N., Rohde K., Vaisman M., Kineman R.D., Frohman L.A.: Isolated familial somatotropinomas: establishment of linkage to chromosome 11q13.1-11q13.3 and evidence for a potential second locus at chromosome 2p16-12.J. Clin. Endocrinol. Metab. 2000; Feb.85(2):707-14.
Gaztambide S., Vazquez .F, Castaño L. : Diagnosis and treatment of multiple endocrine neoplasia type 1 (MEN1). Minerva Endocrinol. 2013: Mar. 38(1), 17-28.
Imai T., Uchino S., Okamoto T., Suzuki S., Kosugi S., Kikumori T., Sakurai A. : High penetrance of pheochromocytoma in multiple endocrine neoplasia 2 caused by germ line RET codon 634 mutation in Japanese patients. Eur. J. Endocrinol. 2013: Apr. 15; 168(5), 683-7.
Kolačkov K., Tupikowski K., Bednarek-Tupikowska G. : Genetic aspects of pheochromocytoma. Adv. Clin. Exp. Med. 2012: Nov-Dec 21(6), 821-9.
Lu Y.Y., Zhu F., Jing D.D., Wu X.N., Lu L.G., Zhou G.Q., Wang X.P. : Multiple endocrine neoplasia type 1 with upper gastrointestinal hemorrhage and perforation: a case report and review. World J. Gastroenterol. 2013: Feb. 28; 19(8), 1322-6.
Öberg K. : The genetics of neuroendocrine tumors. Semin. Oncol. 2013: Feb. 40(1), 37-44.
Purri P., Zotti G.C., Fimmanò A , Liguori G., Della Volpe N. : Gli apudomi della sfera bilio‑duodeno‑pancreatica.Comun. III Congr. Naz. Soc. It. Endocrinochirurgia. Padova, 1983. Antol. Med. It.: IV; 1984 (atti del congresso).
Qari F. : RET codon 618 mutations in Saudi families with multiple endocrine neoplasia Type 2A and familial medullary thyroid carcinoma. Ann. Saudi Med. 2013: Mar-Apr;33(2), 155-8.
Sala E., Ferrante E., Verrua E., Malchiodi E., Mantovani G., Filopanti M., Ferrero S., Pietrabissa A., Vanoli A., La Rosa S., Zatelli M.C., Beck-Peccoz P., Verga U. : Growth hormone-releasing hormone-producing pancreatic neuroendocrine tumor in a multipleendocrine neoplasia type 1 family with an uncommon phenotype. Eur. J. Gastroenterol. Hepatol. 2013: Mar 29.
Salehian B., Samoa R. : RET gene abnormalities and thyroid disease: who should be screened and when. J. Clin. Res. Pediatr. Endocrinol. 2013:5 ; Suppl. 1,70-8.
Salmela P.I. : Multiple endocrine neoplasia type 1. Duodecim. 2012: 128(22), 2345-54.
Soares B.S., Frohman L.A.: Isolated familial somatotropinoma. Pituitary 2004;7(2):95-101.
Thakker R.V. : Multiple endocrine neoplasia type 1. Indian J. Endocrinol. Metab. 2012: Dec. 16 (Suppl 2), 272-4.
Twigt B.A., Scholten A., Valk G.D., Rinkes I.H., Vriens M.R. : Differences between sporadic and MEN related primary hyperparathyroidism; clinical expression, preoperative workup, operative strategy and follow-up. Orphanet J. Rare Dis. 2013: Apr. 1, 8-50.
Zotti G.C., Purri P., Masciariello S., Aceto A , Santini M., Di Crescenzo V.G. : Gastropatie acute ed endocrinopatie. Considerazioni patogenetiche, diagnostiche e terapeutiche. La Riforma Medica 1982; 97, 491.
Zotti G.C., Purri P., Santaniello W., Pisani R. : Le lesioni gastriche da tumori del sistema APUD. La Riforma Medica 1984; 99, 393.